Introduction
Lung Adenocarcinoma (LUAD) is the most common histological subtype of non-small cell lung cancer (NSCLC), accounting for
about 40% of lung cancer [1]. With the rapid advances in diagnosis and treatment like surgery, radiotherapy, and molecular
therapy, there is a remarkable improvement on clinical outcomes
of patients with LUAD. However, the five-year Overall Survival
(OS) of LUAD still remains lower than 20% [2]. There are proofs
demonstrated that the exploration and application of molecular
biomarkers can provide prognosis value for LUAD [3].
Spermatogenesis-Associated Serine-Rich 2-Like Gene (SPAT-S2L), which is ubiquitously expressed in multiple tissues [4], has
the greatest number of GenBank accessions belonging to the lung
with its function remaining elusive according to data collected by
Ace View tool [5,6]. Whole genome association analysis revealed
that SPATS2L is an important response gene to bronchodilators
for asthma patients, and SPATS2L was reported to be associated
with the therapy response of children with asthma [7,8]. Its over-expression is related to the carcinogenesis of glioma and hepatocellular carcinoma [6,9]. Another study discovered a relationship
between the susceptibility loci of rs985256-SPATS2L and the susceptibility of Coal Workers’ Pneumoconiosis (CWP) [10].
Based on the studies mentioned above, there seems to be a
certain correlation between SPATS2L and the lungs. However,
there is almost no research on the role of SPATS2L in the pathogenesis, development, and prognosis of lung adenocarcinoma.
Therefore, we speculate that SPATS2L is a potential biomarker for
lung adenocarcinoma. In this study, we first explored the differential expression of SPATS2L mRNA in LUAD tissues and normal lung
tissues and analyzed the correlation between SPATS2L expression
and SPATS2L DNA methylation based on The Cancer Genome Atlas
(TCGA) LUAD dataset. The prognostic significance of SPATS2L expression and DNA methylation was also evaluated by meta-analysis. Tumor Immune Estimation Resource (TIMER) database was
adopted to assess the potential correlation between SPATS2L and
immune infiltration cells in LUAD. Then we analyzed the biological
process of SPATS2L participating in LUAD through GO enrichment
analysis. All of these works were intended to evaluate the prognosis significance of SPATS2L methylation and mRNA expression for
LUAD patients and its latent value as a biomarker of LUAD.
Patients and methods
Mining data from public databases
GEPIA, an online gene expression profile interactive analysis
website (http://gepia.cancer-pku.cn/index.html), was adopted to
study the differential expression of SPATS2L mRNA in LUAD tissues
and normal tissues. The clinical data, transcription, and methylation profiles of LUAD patients were collected from two articles
[11,12], and the TCGA database (https://www.cancer.gov/tcga).
Patients with complete clinical and transcriptional data were included. Additionally, we further verified the prognostic role of
SPATS2L in LUAD by analyzing clinical data and SPATS2L mRNA expression data from 18 articles and TCGA database. A total of 2757
patients diagnosed with LUAD were brought into this analysis.
Meta-analysis
A systematic search of PubMed, Web of Science, and Embase databases was conducted to identify all published studies on the
association between SPATS2L expression and the prognosis of
LUAD. We used meta-analysis to analyze data from 1 database and
18 articles to assess the overall prognostic significance of SPATS2L
expression for patients with LUAD. The consolidated HR and 95%
CI were calculated to assess the correlation between SPATS2L expression and prognosis in patients with LUAD. The heterogeneity
of data from 18 articles and a database was determined by the Q
test (I2 statistics). If there’s no significant heterogeneity (I2<50%),
a fixed-effects model would be performed. Otherwise, a random-effects model would be applied. The meta-analysis was accomplished by STATA 15.1 software.
TIMER database analysis
The correlation between SPATS2L expression and the abundance of 7 immune cells (CD4+T cells, CD8+T cells, B cells, neutrophils, myeloid dendritic cells, macrophages, and monocytes) in
LUAD was examined using the TIMER algorithm (https://cistrome.
shinyapps.io/timer/), a robust website able to automatically analyze and visualize the correlation between immune infiltration levels and a range of variables.
GO enrichment analysis
The Gene Ontology analysis was conducted based on the Glio-Vis database (http://gliovis.bioinfo.cnio.es/). The patients with
lung adenocarcinoma in the GlioVis database were initially divided
into high and low SPATS2L expression groups. The false discovery
rate for selecting differentially expressed genes between the two
groups was less than 0.05. Gene terms with | logFC | ≥1 along
with a P value less than 0.05 was considered significant. Then GO
enrichment analysis and biological processes were chosen to explore the functional role of SPATS2L in lung adenocarcinoma.
Statistics
Data was analyzed by the Medcalc program (version 19.4) and
GraphPad Prism (version 6.0). Low and high SPATS2L expression
groups were established based on the median SPATS2L mRNA
expression values in different databases. Similarly, SPATS2L hypomethylation and hypermethylation groups were established
based on the median value of SPATS2L DNA methylation in the TCGA-LUAD database. The relationship between SPATS2L expression
or DNA methylation and a range of classification variables was
analyzed using chi-square or Fisher exact tests. The difference between the continuous indices of the two groups with normal distribution was determined by Student’s t-test, and the continuous
indices with skewed distribution were tested by a nonparametric
test. Spearman rank correlation coefficient and Pearson correlation coefficient were used to measure the correlation between
SPATS2L expression and SPATS2L DNA methylation level. Kaplan
Meier curve was used to evaluate the prognostic significance of
SPATS2L expression and SPATS2L DNA methylation. The P values
on both sides less than 0.05 were statistically significant.
Results
The prognostic value of SPATS2L methylation for LUAD
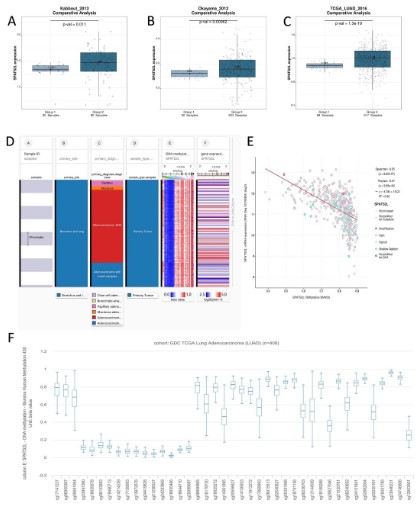
RNA-seq data from Kabbout_2013 (P=0.011), Okayama_2012
(P=0.00062), and TCGA (P<0.0001) showed that SPATS2L mRNA
was highly expressed in LUAD tissues than that in the normal lung tissues (Figure 1A, B and C). As exhibited in the heatmap, SPATS2L
was abundantly expressed in bronchus and lung tissues, and there
was a strong correlation between SPATS2L and adenocarcinoma
(Figure 1D). Besides, SPATS2L mRNA expression was negatively
correlated (r=0.32, P<0.0001) with SPATS2L methylation level
(Figure 1E). Figure 1F exhibits the distribution of 41 SPATS2L CpG
islands. Figure 2A is the heatmap of methylation sites of SPATS2L,
displaying hypomethylated and hypermethylated CpG islands. 8
SPATS2L methylation sites at which the methylation level was determined as most strongly correlated with SPATS2L mRNA expression were selected to assess the prognostic significance of SPATS2L
methylation for patients with LUAD through Kaplan-Meier plots,
manifesting that hypermethylation at 5 of 8 SPATS2L methylation
sites was relevant to a longer survival time (Figure 2B, C, D, E, F, G,
H and I). Based on the pooled HR (95% CI) of 0.92 (0.85, 1.00), we
inferred that the high methylation level of SPATS2L methylation
sites was a protective factor for LUAD OS (Figure 2J).
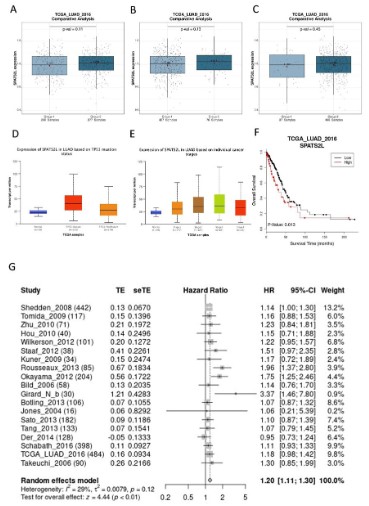
Assessment of SPATS2L differential expression in subgroups
based on TCGA database
The nonparametric test was applied to confirm the expression
difference of SPATS2L mRNA in TCGA database-sourced populations grouped by gender (Figure 3A), IDH1 type (Figure 3B),
MGMT methylation (Figure 3C), TP53 mutation status (Figure
3D), cancer stage (Figure 3E). On the whole, no close relationship
was found between SPATS2L expression in LUAD tissues and gender (P=0.11), IDH1 type (P=0.13), and MGMT methylation level
(P=0.45). According to the large standard deviation gaps between
groups, SPATS2L expression in LUAD was strongly correlated to
TP53 mutation and individual cancer stage. Additionally, Kaplan-Meier curves visualized that low expression of SPATS2L in LUAD
prolonged OS (Figure 3F).
Meta-analysis of SPATS2L expression for patients OS with
LUAD
We performed meta-analysis using the data extracted from
18 articles and 1 database to observe the correlation of SPATS2L
mRNA expression with the OS of LUAD patients (Figure 3G). With
a pooled HR (95% CI) of 1.20 (1.11; 1.03), and no significant heterogeneity among 19 data sources (I2=29%, P=0.12), we concluded that low expression level of SPATS2L mRNA was a protective
factor for LUAD OS.
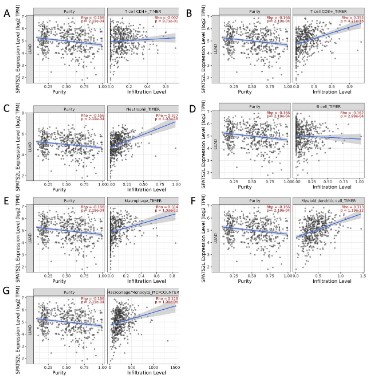
Correlation between SPATS2L mRNA expression and immune
infiltration cells
Spearman correlation analysis was used to evaluate the relationship of SPATS2L mRNA expression with immune cell infiltration by searching the TIMER database (Figure 4). It was illustrated
that the relation between SPATS2L mRNA expression and immune
infiltrating CD4+T cell (ρ=-0.002, P=0.971), CD8+T cell (ρ=0.355,
P<0.05), neutrophil (ρ=0.377, P<0.05), B cell (ρ=-0.162, P<0.05),
macrophage (ρ=0.314, P<0.05), myeloid dendritic cell (ρ=0.313,
P<0.05), and monocyte (ρ=0.218, P<0.05) was not close.
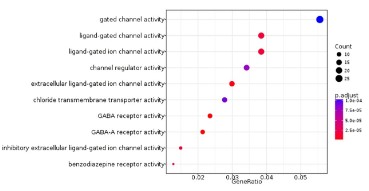
GO analysis of SPATS2L involved in cell function
Gene Ontology analysis based on GlioVis database was applied to explore the cell function in which SPATS2L participated.
As displayed in Figure 5, SPATS2L mainly engaged in cell functions
like gated channel activity, ligand-gated channel activity, ligand-gated ion channel activity, channel regulator activity, extracellular
ligand-gated ion channel activity, chloride transmembrane transporter activity. Previous studies revealed that low expression of
Cystic Fibrosis Transmembrane Regulator (CFTR) was remarkedly
related to the development of non-small cell lung cancer and tumor invasion ability [13-15]. Hence, SPATS2L was assumed to affect LUAD development indirectly by regulating channel activity
like chloride transmembrane transporter activity.
Discussion
In recent years, lung cancer has become the most common
malignant tumor, posing a serious threat to global human health,
with an annual incidence rate of 7.5% [16]. As the major histological subtype of NSCLC, LUAD causes more than 10000 deaths
worldwide each year [17]. Besides, the apparent heterogeneity
of LUAD makes its diagnosis and treatment difficult [18]. A large
number of literature studies have shown that environmental factors, as well as genetic and epigenetic factors, can affect the occurrence and development of lung adenocarcinoma [19-23]. Epigenetic studies of lung cancer illustrated that there are abnormal methylation states in various lung cancer patient samples, such as
sputum [24], bronchoalveolar lavage [25], and cancer tissue [26].
Furthermore, many tumor related genes, including oncogenes
and tumor suppressor genes, change their methylation status in
the early stages of lung cancer [27]. DNA methylation can be used
to track the recurrence of early lung adenocarcinoma (LUAD) after
surgery [28]. Therefore, altered methylation status can be used in
lung oncology to identify biomarkers to assist in tumor detection
and predict cancer prognosis.
In this study, we investigated and validated the prognostic
value of SPATS2L methylation in patients with LUAD. After analyzing the data obtained from TCGA, we found for the first time
that the expression of SPATS2L in lung adenocarcinoma tissue was
significantly higher than that in normal lung tissues, which was verified by the data obtained from the other two articles. In addition, we statistically concluded that the mRNA level of SPATS2L
was negatively correlated with the methylation level of CpG island. Among the 8 CpG islands most related to the mRNA expression level of SPATS2L, the hypermethylation level of 5 of them was
related to a longer survival time. Meta-analysis results indicated
that the hypermethylation of all CpG islands was a protective factor for the prognosis of LUAD. To better explain the relationship
between SPATS2L methylation level and the prognosis of LUAD,
we conducted a meta-analysis of LUAD patients’ data from the
TCGA database and 18 articles. The results indicated that a high
SPATS2L methylation level was a protective factor for the prognosis of LUAD, that is, low SPATS2L expression prolonged the OS
of LUAD patients. Likewise, SPATS2L expression is upregulated in
patients with glioma, and its low expression is involved in a better OS [9]. For patients with acute myeloid leukemia, high expression of SPATS2L leads to a significantly lower survival rate than
those with low expression [29]. Zhao et al. proposed a predictive
model that combined SPATS2L and 9 other genes to evaluate the
prognosis of acute myeloid leukemia, demonstrating an inferior
5-year OS in high-risk patients [30]. SPATS2L was also reported to
be involved in mental disease [31], systemic lupus erythematosus
[32], asthma [33], etc. Though studies on SPATS2L are comparatively fewer at present, it is clear that as an oncogene, upregulation of SPATS2L can lead to many diseases. While the specific role
of SPATS2L in LUAD still needs to be deeply explored.
Immune cells infiltrated in the tumor microenvironment were
proven to mediate the efficacy of immunotherapy, which is a
promising target in drug development [34,35]. The increase in the
proportion of effector cells in the immune microenvironment often indicates a better prognosis for patients [36,37]. This study
analyzed the relationship between SPATS2L and LUAD immune
infiltrating cells through the TIMER database. Unfortunately, no
robust correlation was found between SPATS2L and immune infiltration cells.
GO enrichment analysis manifested that SPATS2L was closely
related to the activity of various channels on the cell membrane,
including the chlorine transport transporter. Coincidentally, the
Crystal Fibrosis Transmembrane Regulator (CFTR) is a cAMP-dependent chloride ion channel that regulates the growth and proliferation of tumor cells in various cancers [38-41]. Studies have
shown that the expression of CFTR is downregulated in lung cancer [13-15]. Downregulation of CFTR expression promotes EMT
progression and metastasis of lung adenocarcinoma cell line A549
by upregulating the uPA/uPAR system. Inhibiting the functional
activity of CFTR chloride channels also promotes the EMT process
of A549 and increases the migration and invasion ability of lung
cancer cells [15]. Therefore, SPATS2L may affect the development
of lung adenocarcinoma by regulating the expression or function
of CFTR. Given that high methylation of SPATS2L is a protective
factor for LUAD, we speculate that low expression of SPATS2L
could upregulate CFTR, thereby slowing down the migration and
invasion ability of lung cancer cells. However, this conjecture
needs further research to prove.
Conclusions
In summary, the expression of SPATS2L was upregulated in
LUAD. High methylation and low mRNA expression of SPATS2L were associated with better OS of patients with LUAD. SPATS2L
perhaps plays an important role in the occurrence and development of LUAD, and is a promising prognostic marker for LUAD,
bringing more possibilities for improving the prognosis of patients.
Acknowledgements: This study benefited from the Cancer Genome Atlas (TCGA) database, Tumor Immune Estimation Resource
(TIMER) and GlioVis database. We appreciate the data platform
and the authors uploaded their data.
References
- Chen Z, Fillmore CM, Hammerman PS, et al. Non-small-cell lung
cancers: A heterogeneous set of diseases. Nature reviews Cancer.
2014; 14: 535-46.
- Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. The New England
journal of medicine. 2012; 366: 2443-54.
- Jiao G, Wang B. NK Cell Subtypes as Regulators of Autoimmune Liver Disease. Gastroenterology research and practice. 2016; 2016:
6903496.
- Fagerberg L, Hallström BM, Oksvold P, et al. Analysis of the human
tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Molecular & cellular
proteomics: MCP. 2014; 13: 397-406.
- Thierry-Mieg D, Thierry-Mieg J. AceView: A comprehensive cDNA-supported gene and transcripts annotation. Genome biology.
2006; 7: S12.1-14.
- Min P, Li W, Zeng D, et al. A single nucleotide variant in microRNA-1269a promotes the occurrence and process of hepatocellular carcinoma by targeting to oncogenes SPATS2L and LRP6. Bulletin du
cancer. 2017; 104: 311-320.
- Himes BE, Jiang X, Hu R, et al. Genome-wide association analysis
in asthma subjects identifies SPATS2L as a novel bronchodilator response gene. PLoS genetics. 2012; 8: e1002824.
- Tse SM, Krajinovic M, Chauhan BF, et al. Genetic determinants of
acute asthma therapy response in children with moderate-to-severe asthma exacerbations. Pediatric pulmonology. 2019; 54: 378-385.
- Wang H, Wang X, Xu L, et al. Analysis of the EGFR Amplification and
CDKN2A Deletion Regulated Transcriptomic Signatures Reveals the
Prognostic Significance of SPATS2L in Patients With Glioma. Frontiers in oncology. 2021; 11: 551160.
- Wang T, Sun W, Wu H, et al. Respiratory traits and coal workers’
pneumoconiosis: Mendelian randomisation and association analysis. Occupational and environmental medicine. 2021; 78: 137-141.
- Kabbout M, Garcia MM, Fujimoto J, et al. ETS2 mediated tumor
suppressive function and MET oncogene inhibition in human non-small cell lung cancer. Clinical cancer research : An official journal
of the American Association for Cancer Research. 2013; 19: 3383-95.
- Okayama H, Kohno T, Ishii Y, et al. Identification of genes upregulated in ALK-positive and EGFR/KRAS/ALK-negative lung adenocarcinomas. Cancer research. 2012; 72: 100-11.
- Li Y, Sun Z, Wu Y, et al. Cystic fibrosis transmembrane conductance
regulator gene mutation and lung cancer risk. Lung cancer (Amsterdam, Netherlands). 2010; 70: 14-21.
- Tian F, Zhao J, Fan X, et al. Weighted gene co-expression network
analysis in identification of metastasis-related genes of lung squamous cell carcinoma based on the Cancer Genome Atlas database.
Journal of thoracic disease. 2017; 9: 42-53.
- Li J, Zhang JT, Jiang X, et al. The cystic fibrosis transmembrane conductance regulator as a biomarker in non-small cell lung cancer.
International journal of oncology. 2015; 46: 2107-15.
- Jemal A, Bray F, Center MM, et al. Global cancer statistics. CA: A
cancer journal for clinicians. 2011; 61: 69-90.
- Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics
2018: GLOBOCAN estimates of incidence and mortality worldwide
for 36 cancers in 185 countries. CA: A cancer journal for clinicians.
2018; 68: 394-424.
- Xu F, He L, Zhan X, et al. DNA methylation-based lung adenocarcinoma subtypes can predict prognosis, recurrence, and immunotherapeutic implications. Aging. 2020; 12: 25275-25293.
- Gong H, Li Y, Yuan Y, et al. EZH2 inhibitors reverse resistance to
gefitinib in primary EGFR wild-type lung cancer cells. BMC cancer.
2020; 20: 1189.
- Fabrizio FP, Mazza T, Castellana S, et al. Epigenetic Scanning of
KEAP1 CpG Sites Uncovers New Molecular-Driven Patterns in Lung
Adeno and Squamous Cell Carcinomas. Antioxidants (Basel, Switzerland). 2020; 9.
- Wang J, He L, Tang Y, et al. Development and validation of a nomogram with an epigenetic signature for predicting survival in patients with lung adenocarcinoma. Aging. 2020; 12: 23200-23216.
- Huang J, Zhang Q, Shen J, et al. Multi-omics analysis identifies potential mechanisms of AURKB in mediating poor outcome of lung
adenocarcinoma. Aging. 2021; 13: 5946-5966.
- Shi R, Bao X, Unger K, et al. Identification and validation of hypoxia-derived gene signatures to predict clinical outcomes and
therapeutic responses in stage I lung adenocarcinoma patients.
Theranostics. 2021; 11: 5061-5076.
- Shivapurkar N, Stastny V, Xie Y, et al. Differential methylation of a
short CpG-rich sequence within exon 1 of TCF21 gene: A promising cancer biomarker assay. Cancer epidemiology, biomarkers &
prevention: A publication of the American Association for Cancer
Research, cosponsored by the American Society of Preventive Oncology. 2008; 17: 995-1000.
- Schmidt B, Liebenberg V, Dietrich D, et al. SHOX2 DNA methylation
is a biomarker for the diagnosis of lung cancer based on bronchial
aspirates. BMC cancer. 2010; 10: 600.
- Han W, Wang T, Reilly AA, et al. Gene promoter methylation assayed in exhaled breath, with differences in smokers and lung cancer patients. Respiratory research. 2009; 10: 86.
- Belinsky SA. Gene-promoter hypermethylation as a biomarker in
lung cancer. Nature reviews Cancer. 2004; 4: 707-17.
- Brock MV, Hooker CM, Ota-Machida E, et al. DNA methylation
markers and early recurrence in stage I lung cancer. The New England journal of medicine. 2008; 358: 1118-28.
- Zhang Y, Xiao L. Identification and validation of a prognostic 8-gene
signature for acute myeloid leukemia. Leukemia & lymphoma.
2020; 61: 1981-1988.
- Zhao Y, Niu LT, Hu LJ, et al. Comprehensive analysis of ECHDC3 as a
potential biomarker and therapeutic target for acute myeloid leukemia: Bioinformatic analysis and experimental verification. Frontiers in oncology. 2022; 12: 947492.
- Smeland OB, Wang Y, Frei O, et al. Genetic Overlap Between
Schizophrenia and Volumes of Hippocampus, Putamen, and Intracranial Volume Indicates Shared Molecular Genetic Mechanisms.
Schizophrenia bulletin. 2018; 44: 854-864.
- Chen H, Huang L, Jiang X, et al. Establishment and analysis of a disease risk prediction model for the systemic lupus erythematosus
with random forest. Frontiers in immunology. 2022; 13: 1025688.
- Sordillo JE, McGeachie M, Lutz SM, et al. Longitudinal analysis of
bronchodilator response in asthmatics and effect modification of
age-related trends by genotype. Pediatric pulmonology. 2019; 54:
158-164.
- Liu X, Hogg GD, DeNardo DG. Rethinking immune checkpoint
blockade: ‘Beyond the T cell’. Journal for immunotherapy of cancer. 2021; 9.
- Dafni U, Michielin O, Lluesma SM, et al. Efficacy of adoptive therapy with tumor-infiltrating lymphocytes and recombinant interleukin-2 in advanced cutaneous melanoma: A systematic review and
meta-analysis. Annals of oncology: Official journal of the European
Society for Medical Oncology. 2019; 30: 1902-1913.
- Tanaka A, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell research. 2017; 27: 109-118.
- Savas P, Salgado R, Denkert C, et al. Clinical relevance of host immunity in breast cancer: From TILs to the clinic. Nature reviews
Clinical oncology. 2016; 13: 228-41.
- Sun TT, Wang Y, Cheng H, et al. Disrupted interaction between
CFTR and AF-6/afadin aggravates malignant phenotypes of colon
cancer. Biochimica et biophysica acta. 2014; 1843: 618-28.
- Tu Z, Chen Q, Zhang JT, et al. CFTR is a potential marker for nasopharyngeal carcinoma prognosis and metastasis. Oncotarget.
2016; 7: 76955-76965.
- Xie C, Jiang XH, Zhang JT, et al. CFTR suppresses tumor progression through miR-193b targeting urokinase plasminogen activator
(uPA) in prostate cancer. Oncogene. 2013; 32: 2282-91- 2291.e1-7.
- Xu J, Yong M, Li J, et al. High level of CFTR expression is associated
with tumor aggression and knockdown of CFTR suppresses proliferation of ovarian cancer in vitro and in vivo. Oncology reports.
2015; 33: 2227-34.