Introduction
Pituitary metastases are rare, accounting for approximately 1-3.6% of all surgically treated sellar lesions, but are increasingly recognized due to improved survival in cancer patients and advancements in imaging techniques and surveillance cancer pathways [1,2]. Their diagnosis poses significant challenges due to their radiological similarity to primary pituitary adenomas and often a non-specific clinical presentation [3]. Although breast and lung origin metastases are the most common in the pituitary gland, metastases from gastrointestinal and neuroendocrine tumors—including pancreas, thyroid, and colon—have also been documented, albeit far less frequently [3-9].
In the majority of cases, pituitary metastases remain clinically silent and thus undiagnosed. Literature has shown pituitary involvement in up to 28% of cancer patient autopsies, though the symptomatic cases remain infrequent [3,10]. In addition, metastatic Neuroendocrine Tumors (NETs) to the pituitary are even rarer but clinically significant contributors, occasionally presenting as “collision tumors» within preexisting pituitary adenomas, often diagnosed intraoperatively or on histology [9,11,12]. Importantly, up to 44% of pituitary metastases are the initial manifestation of a previously undiagnosed malignancy, underscoring the need for high clinical suspicion [5].
While pituitary metastases are a challenge, as described, their combination with a giant pituitary lesion can complicate management. Giant pituitary adenomas constitute 6-15% of all pituitary lesions and pose substantial surgical and endocrinological challenges due to their size, invasiveness, and proximity to critical neurovascular structures [13,14]. These tumors often result in mass effect symptoms—visual impairment, headaches, cranial nerve palsies—as well as hypopituitarism, particularly when extending suprasellarly or laterally into the cavernous sinus [15,16].
In this study, we present a unique case of a giant pituitary metastasis from a duodenopancreatic neuroendocrine tumor (GEPNETs), initially mistaken for a primary pituitary adenoma. We also aim to contextualize its clinical features, diagnostic process, and therapeutic considerations through a focused review of the existing literature. By highlighting both individual and collective experiences, we seek to improve recognition of this rare entity and support more informed clinical decision-making.
Methods
Due to this case of giant pituitary metastasis from a duodenopancreatic NET that we encountered, we conducted a literature review following PRISMA guidelines to identify similar reported cases of pituitary metastases originating from gastrointestinal or pancreatic neuroendocrine tumors (GEP-NETs). We searched the PubMed database for articles published up to April 2025 using the terms: “pituitary metastasis,” “neuroendocrine tumor,” “pancreatic,” “duodenal,” “gastrointestinal,” and “sellar mass.”
Eligibility criteria
Included studies should fulfil the following criteria: (1) English language case reports, case series, or systematic reviews, (2) adults or children, (3) pathologically confirmed pituitary metastases from a gastrointestinal or pancreatic NET, and (4) articles providing clinical, imaging, and treatment data.
Exclusion criteria
Excluded studies will be those (1) non-metastatic pituitary lesions, (2) non-gastrointestinal or pancreatic NET sources of pituitary metastasis, (3) colon NET sources of metastasis, and (3) Studies lacking patient level data or histological confirmation. Finally, (4) papers published in languages other than English will be excluded from this review.
Selection process
The study selection will follow a double-reviewer process. The records identified in PubMed were checked for duplicates and screened according to the selection criteria. The retrieved papers were examined in full for eligibility. Discrepancies were resolved through consensus.
Data extraction G synthesis
Data extracted included patient demographics, primary tumor origin, presenting symptoms, hormonal profile, imaging findings, surgical approach, histopathology, and outcomes. However, while a structured search strategy was applied to identify relevant studies on pituitary metastases from GEP-NETs, the nature of the available data does not allow its classification as a formal systematic review. Most published reports are isolated case studies or small series with heterogeneous reporting of clinical variables, imaging findings, and treatment outcomes. Therefore, this work is best considered a literature review, aiming to synthesize published knowledge, highlight diagnostic challenges, and guide future recognition and management of this rare clinical entity.
Results
Case presentation
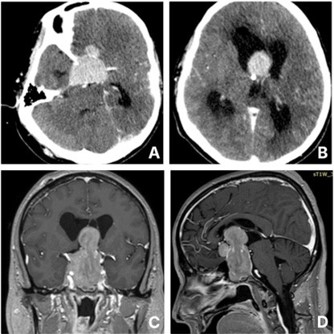
A 57-year-old male with no significant past medical history presented to the emergency department with a sudden onset of severe headache, vomiting, and progressive loss of consciousness. He was intubated for airway protection on arrival due to collapse. Initial CT imaging revealed a giant sellar lesion extending into the third ventricle, causing obstructive hydrocephalus (Figure 1). An emergent External Ventricular Drainage (EVD) was placed, and the patient was admitted to the Intensive Care Unit (ICU), where he was started on intravenous dexamethasone. Further systemic examination revealed an absence of body hair on the trunk and limbs, while facial and pubic hair remained intact. He exhibited no gynecomastia or galactorrhoea, and testicular volume was within normal limits. The family reported long-standing hair loss since adolescence, but had fathered four children by the age of 30. Laboratory investigations showed low levels of Follicle-Stimulating Hormone (FSH), Luteinizing Hormone (LH), testosterone, Growth Hormone (GH), and Free Thyroxine (FT4), with a normal Thyroid Stimulating Hormone (TSH) level and low Prolactin (PRL). He was started on levothyroxine for presumed central hypothyroidism.
MRI confirmed the presence of a giant sellar lesion extending into the suprasellar region and 3rd ventricle to the foramen of Monro (Figure 1). The lesion exhibited enhancement and diffusion restriction on Susceptibility-Weighted Imaging (SWI), with invasion of the bilateral cavernous sinuses and encasement of the internal carotid arteries. Also, the optic chiasm was compressed. In reduced sedation attempts, the patient was decerebrating.
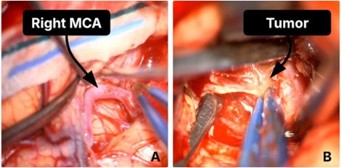
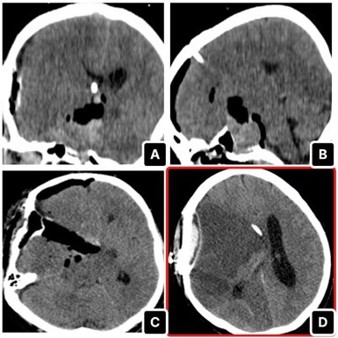
Given the lesion’s size and intraventricular extension, the patient underwent pterional craniotomy for debulking as the first step of tumor excision (Figure 2). shows the tumour debulking and the identification of the right Middle Cerebral Artery (MCA). As the MCA was promptly identified during the operation, local irrigation with papaverine solutions was applied. Postoperatively, the patient was transferred again to the ICU, and CT head showed the extent of debulking (Figure 3). Two days post-op, the patient developed anisocoria, and the repeated CT scan showed a right-sided MCA infarct with extensive midline shift (Figure 3). No matter the intra-operative papaverine irrigation, MCA vasospasm was quite evident. Therefore, we did not continue with further endoscopic transsphenoidal resection. The patient subsequently succumbed to complications.
Microscopically, the material consists of fragments of a cellular neoplasm with a predominantly nested growth pattern and a prominent vascular layer. The neoplastic cells in several locations are arranged around thin-walled, papillary vessels. The neoplastic cells are characterized by eosinophilic cytoplasm and relatively uniform nuclei with finely granular chromatin. ΑΕ1/ΑΕ3 (+), INSM1 (+), GARA3 (+), CK19 (+), ISLE1 (+), 2-3 p53, Ki67 8%. Brain infiltrate from a NET G2. Histopathological examination of the tumor revealed features consistent with a neuroendocrine neoplasm (NET G2), with a presumed primary origin in the duodenum or pancreas.
Review results
A total of 23 unique cases of pituitary metastasis from pancreatic or gastrointestinal neuroendocrine tumors (GEP-NETs) were identified. These cases primarily involved pancreatic primaries, followed by intestinal and duodenal NETs. Visual symptoms, anterior hypopituitarism, and non-specific neurological complaints were the most common presenting features. Imaging frequently revealed suprasellar extension and cavernous sinus invasion, mimicking invasive pituitary adenoma. In all cases, the final diagnosis was established via postoperative histopathological analysis.
Most cases are isolated reports or small series lacking standardized outcome measures, consistent imaging protocols, or uniform diagnostic criteria. Many publications omit critical details such as hormonal profiles, treatment, or follow-up outcomes. Furthermore, the heterogeneity in reporting styles and the absence of comparative data prevent this study from being classified as a systematic review. Therefore, this work constitutes a structured literature review, aimed at synthesizing the current knowledge and highlighting the diagnostic and management challenges associated with this rare clinical entity.
Discussion
The presented case highlights a highly unusual presentation of a giant pituitary metastasis originating from a duodenopancreatic neuroendocrine tumor—an entity that remains extremely rare in the literature. While the pituitary gland is a generally infrequent site of metastasis, the co-occurrence of a large sellar mass and the absence of a known primary malignancy at presentation complicate the clinical picture significantly. The initial radiological and endocrinological features in our patient mimicked those of a benign pituitary macroadenoma, underscoring the difficulty of preoperative diagnosis. Only after surgical excision, the histopathological analysis provided the definitive confirmation of metastatic neuroendocrine tumor in the pituitary.
Molecular background
Molecularly, pituitary metastasis is largely influenced by the tumor microenvironment, where vascular factors, hormone gradients, and immune infiltrates influence tumor seeding and proliferation (1). For example, prolactin-rich settings may draw breast cancer cells to the pituitary, while Vascular Endothelial Growth Factor (VEGF) and hypoxic gradients can promote implantation (1). Recent studies have demonstrated the oncogenic role of the Pituitary Tumor-Transforming Gene 1 (PTTG1) in promoting tumor invasiveness and metastasis across gastrointestinal malignancies, suggesting new possibilities for targeted diagnosis and treatment [17-19]. PTTG1 oncogene is found overexpressed in 60-66% of esophageal squamous cell carcinomas, correlating strongly with lymph node metastases and decreased survival [17,18]. The same marker of tumor aggressiveness has been observed in 65.3% of gastric carcinomas and is associated with metastasis [18,19].
Additionally, genetic syndromes such as MEN1 and MEN4, associated with mutations in MEN1 and CDKN1B, respectively, suggest a shared oncogenic pathway in endocrine and gastrointestinal malignancies [20-23]. Furthermore, historical studies have observed the loss of heterozygosity at chromosome 13q12-14, involving tumor suppressors such as RB1 and BRCA2, in 16% of parathyroid adenomas and some rare pituitary tumors, raising thoughts of a shared genetic vulnerability across endocrine neoplasms [21]. Lastly, pancreatic tumors that often express chromogranin A, synaptophysin, and Ki-67 can be differentiated histopathologically from primary pituitary adenomas [1,4].
Clinical characteristics
Clinical presentation of pituitary metastases varies based on tumor characteristics and biological behavior. Recent studies revealed that pituitary metastases often present with headache (67%), visual disturbances (86%), anterior hypopituitarism (71%), and diabetes insipidus (38%), the latter reflecting posterior lobe involvement due to its direct systemic blood supply [1,5,24-26]. Symptoms may mimic those of benign pituitary adenomas, complicating early diagnosis. Notably, diabetes insipidus remains the hallmark symptom and can aid in differentiating metastasis from benign adenomas [5]. In some rapidly developed cases, the mass effect was the reason for the presentation [27]. Given that over half of pituitary metastases are only diagnosed intraoperatively, diagnostic difficulties are further underlined [3].
MRI is the preferred imaging modality for diagnosing pituitary lesions. Radiologically, pituitary metastases often mimic adenomas with typical features of pituitary stalk thickening, sellar masses with suprasellar extension, homogeneous contrast enhancement, and cavernous sinus invasion [1,6,10]. Sometimes it can be suspected due to irregular margins and rapid tumor growth, especially in the absence of a known primary sellar lesion [7,24,28]. In fact, up to 24% of patients are diagnosed with pituitary metastasis before their primary malignancy is identified [24]. In rare cases, metastases can also be mistaken for aneurysms, further complicating the diagnosis [25].
Treatment strategies
Therapeutic management of pituitary metastases, particularly those from Neuroendocrine Tumors (NETs), requires a multimodal approach tailored to disease extent and tumor origin. Surgical resection, often via transsphenoidal route, is primarily used for symptom control, especially in cases of visual compromise or significant mass effect, and tissue diagnosis, with subtotal resection achieved in 76% of cases [24,25,27]. However, in cases involving extensive lateral extension, encasement of the internal carotid arteries, or fibrous tumor consistency, transcranial or staged combined approaches may be necessary [14,29]. For giant pituitary lesions similar to the current case, large case-series studies showed that gross total resection was achieved in 63.8% of cases, with postoperative visual improvement in nearly 79% of patients [15]. When anatomical complexity or tumor consistency precludes complete resection, partial debulking can still yield substantial symptom relief.
As sellar metastases often indicate an advanced systemic disease, surgery alone is rarely curative. Adjuvant radiation -including stereotactic radiosurgery and external beam therapyis frequently administered in about 67% of patients and leads to partial symptomatic relief [2,24]. Systemic therapy remains critical, particularly for patients with widespread disease or poor surgical candidacy, with median overall survival ranging from 12 to 25 months depending on histology and intervention [2,24,28]. For pituitary metastases from gastric cancer, regimens including oxaliplatin and fluoropyrimidines provide temporary disease control [7]. For the treatment of hypopituitarism and diabetic insipidus, hormonal replacement is essential. While dopamine agonists have demonstrated usefulness in regulating pancreatic polypeptide levels (Pathak et al., 2004), somatostatin analogs such as octreotide successfully decrease hormone release in NET-related instances [12,30]. Additionally, targeted therapies -including imatinib for GIST metastases and tyrosine kinase inhibitors like sorafeniboffer promising avenues for systemic control [26,27].
Histopathology
In order to distinguish metastases from primary pituitary adenomas, histopathological examination is essential [4,24]. The gastrointestinal origin was confirmed by reported examples of gastric cancer metastases to the pituitary that tested positive for CK7, CK20, CDX-2, and chromogranin A [7,10]. A high Ki-67 index also suggests an aggressive neoplastic process. Similarly, pituitary adenomas have been linked to neuroendocrine tumors that arise in MEN1 disease, such as thymic carcinoids, which raises concerns regarding hereditary risk [23]. Histologically, differentiating pituitary neuroendocrine tumors from metastatic neuroendocrine cancers can be challenging and often requires a complex panel of immunostains and correlation with primary tumor histology [31].
Prognosis
The overall prognosis of pituitary metastasis patients is poor, with a 71% mortality rate during a mean follow-up of 2.4 years [3]. More specifically, the prognosis for patients with gastric cancer metastasizing to the pituitary remains poor. Mortality is frequently due to broad metastatic disease rather than pituitary involvement alone, and reported survival rates are usually less than 12 months after diagnosis [10]. Crucially, surgical resection was associated with improved survival, particularly in patients under 60 and those with primaries other than lung or breast [2]. In contrast, patients with non-solitary pituitary metastasis or no local therapy had significantly worse survival outcomes [28].
Conclusion
To conclude, a rare case of giant pituitary metastasis from a duodenopancreatic neuroendocrine tumor is presented that was initially mistaken for a pituitary adenoma. Diagnostic difficulties persist due to the clinical and radiological similarities between primary pituitary tumors and metastases, especially when pituitary involvement occurs before the original cancer is discovered. The current case highlights how crucial it is to consider metastatic disease in the differential diagnosis of large or atypical sellar masses, particularly in cases of rapid neurological or endocrine profile deterioration. Histopathology is essential for a conclusive diagnosis. Given the typically poor prognosis despite intervention, timely diagnosis and a multidisciplinary approach are critical for optimizing outcomes and guiding appropriate therapeutic strategies.
Declarations
Acknowledgments: The authors declare that there are no acknowledgements to disclose.
Funding: This research did not receive any specific grant from funding agencies in the public, commercial, or notfor-profit sectors.
References
- Kameda-Smith MM, Zhang E, Lannon M, Algird A, Reddy K, Lu JQ. Pituitary metastasis: from pathology to clinical and radiological considerations. J Clin Neurosci. 2021; 93: 231–40.
- Patel KR, Zheng J, Tabar V, Cohen MA, Girotra M. Extended survival after surgical resection for pituitary metastases: clinical features, management, and outcomes of metastatic disease to the sella. Oncologist. 2020; 25: e789–97.
- Sitoci-Ficici KH, Sippl C, Prajsnar A, Saffour S, Linsler S. Sellar metastasis: a rare intraoperative finding—surgical treatment, strategies and outcome. Clin Neurol Neurosurg. 2024; 241: 108280.
- Goulart CR, Upadhyay S, Ditzel Filho LFS, Beer-Furlan A, Carrau RL, Prevedello LM, et al. Newly diagnosed sellar tumors in patients with cancer: a diagnostic challenge and management dilemma. World Neurosurg. 2017; 106: 254–65.
- Loto MG, Rogozinski A, Alfieri A, Ballarino MC, Battistone MF, Chervin A, et al. Pituitary metastases: a diagnostic and therapeutic challenge. Medicina (B Aires). 2024; 84: 505–15.
- Shibata Y, Haruki N, Kuwabara Y, Nishiwaki T, Kato J, Shinoda N, et al. Expression of PTTG (pituitary tumor transforming gene) in esophageal cancer. Jpn J Clin Oncol. 2002; 32: 233–7.
- Dou XL, Zhou N, Mai YL, Guan M, Sun Z, Gao X, et al. Gastric cancer with pituitary metastasis presenting as symptomatic secondary adrenal insufficiency: a case report. J Dig Dis. 2017; 18: 369–72.
- Schatz RA, Rockey DC. Gastrointestinal bleeding due to gastrointestinal tract malignancy: natural history, management, and outcomes. Dig Dis Sci. 2017; 62: 491–501.
- Suzuki K, Tahara S, Hattori Y, Teramoto S, Ishisaka E, Inomoto C, et al. Lung adenocarcinoma metastasis within a pituitary neuroendocrine tumor: a case report with review of literature. Endocr J. 2024; 71: 295–303.
- Pozzessere D, Zafarana E, Buccoliero AM, Pratesi C, Fargnoli R, Di Leo A. Gastric cancer metastatic to the pituitary gland: a case report. Tumori. 2007; 93: 217–9.
- Kato KI, Takeshita Y, Misu H, Ishikura K, Kakinoki K, SawadaKitamura S, et al. Duodenal adenocarcinoma with neuroendocrine features in a patient with acromegaly and thyroid papillary adenocarcinoma: a unique combination of endocrine neoplasia. Endocr J. 2012; 59: 791–6.
- Gur C, Lalazar G, Salmon A, Dubiner V, Gross DJ. Metastatic pancreatic neuroendocrine tumor presenting as a pituitary space occupying lesion: a case report. Pituitary. 2008; 11: 293–7.
- Joshi KC, Kolb B, Khalili BF, Munich SA, Byrne RW. Surgical strategies in the treatment of giant pituitary adenomas. Oper Neurosurg (Hagerstown). 2024; 26: 4–15.
- Luzzi S, Giotta Lucifero A, Rabski J, Kadri PAS, Al-Mefty O. The party wall: redefining the indications of transcranial approaches for giant pituitary adenomas in the endoscopic era. Cancers (Basel). 2023; 15.
- Ke D, Xu L, Wu D, Yang S, Liu S, Xie M, et al. Surgical management of giant pituitary adenomas: institutional experience and clinical outcomes of 94 patients. Front Oncol. 2023; 13: 1255768.
- Koylu B, Firlatan B, Sendur SN, Oguz SH, Dagdelen S, Erbas T. Giant growth hormone-secreting pituitary adenomas from the endocrinologist’s perspective. Endocrine. 2023; 79: 545–53.
- Ito T, Shimada Y, Kan T, David S, Cheng Y, Mori Y, et al. Pituitary tumor-transforming 1 increases cell motility and promotes lymph node metastasis in esophageal squamous cell carcinoma. Cancer Res. 2008; 68: 3214–24.
- Yan S, Zhou C, Lou X, Xiao Z, Zhu H, Wang Q, et al. PTTG overexpression promotes lymph node metastasis in human esophageal squamous cell carcinoma. Cancer Res. 2009; 69: 3283–90.
- Wen CY, Nakayama T, Wang AP, Nakashima M, Ding YT, Ito M, et al. Expression of pituitary tumor transforming gene in human gastric carcinoma. World J Gastroenterol. 2004; 10: 481–3.
- Bergman L, Boothroyd C, Palmer J, Grimmond S, Walters M, Teh B, et al. Identification of somatic mutations of the MEN1 gene in sporadic endocrine tumours. Br J Cancer. 2000; 83: 1003–8.
- Pearce SH, Trump D, Wooding C, Sheppard MN, Clayton RN, Thakker RV. Loss of heterozygosity studies at the retinoblastoma and breast cancer susceptibility (BRCA2) loci in pituitary, parathyroid, pancreatic and carcinoid tumours. Clin Endocrinol (Oxf). 1996; 45: 195–200.
- Frederiksen A, Rossing M, Hermann P, Ejersted C, Thakker RV, Frost M. Clinical features of multiple endocrine neoplasia type 4: novel pathogenic variant and review of published cases. J Clin Endocrinol Metab. 2019; 104: 3637–46.
- Hasani-Ranjbar S, Rahmanian M, Ebrahim-Habibi A, Soltani A, Soltanzade A, Mahrampour E, et al. Ectopic Cushing syndrome associated with thymic carcinoid tumor as the first presentation of MEN1 syndrome: report of a family with MEN1 gene mutation. Fam Cancer. 2014; 13: 267–72.
- Hong S, Atkinson JL, Erickson D, Kizilbash SH, Little JT, Routman DM, et al. Treatment outcome of metastasis to the pituitary gland: a case series of 21 patients with pathological diagnosis. Neurosurg Focus. 2023; 55: E13.
- Yang C, Zhang H, Zhang S, Liu L, Ma B, Lou J, et al. Oculomotor paralysis, postorbital pain, and hypopituitarism as first presentations of metastatic gastric cancer in the pituitary flourished by internal carotid aneurysm: a case report. Medicine (Baltimore). 2015; 94: e2317.
- Deng J, Liao X, Cao H. Neuroendocrine tumors in a patient with multiple endocrine neoplasia type 1 syndrome: a case report and review of the literature. Medicine (Baltimore). 2023; 102: e34350.
- O’Halloran PJ, Hannon AM, Bartels C, McCawley N, Agha A, Brett F, et al. Gastrointestinal stromal tumor metastases to the pituitary: a rare entity. Br J Neurosurg. 2017; 31: 603–4.
- Hong S, Atkinson JL, Erickson D, Kizilbash SH, Little JT, Routman DM, et al. Contemporary treatment outcome of metastases to the pituitary gland. World Neurosurg. 2023; 172: e684–94.
- Tang C, Wang JW, Wang P, Zou DW, Wu N. Staged surgery for irregular giant pituitary adenomas: a report of two cases. Oncol Lett. 2023; 25: 118.
- Pathak RD, Tran TH, Burshell AL. A case of dopamine agonists inhibiting pancreatic polypeptide secretion from an islet cell tumor. J Clin Endocrinol Metab. 2004; 89: 581–4.
- Ragni A, Nervo A, Papotti M, Prencipe N, Retta F, Rosso D, et al. Pituitary metastases from neuroendocrine neoplasms: case report and narrative review. Pituitary. 2021; 24: 828–37.